RNA-Seq Alignment
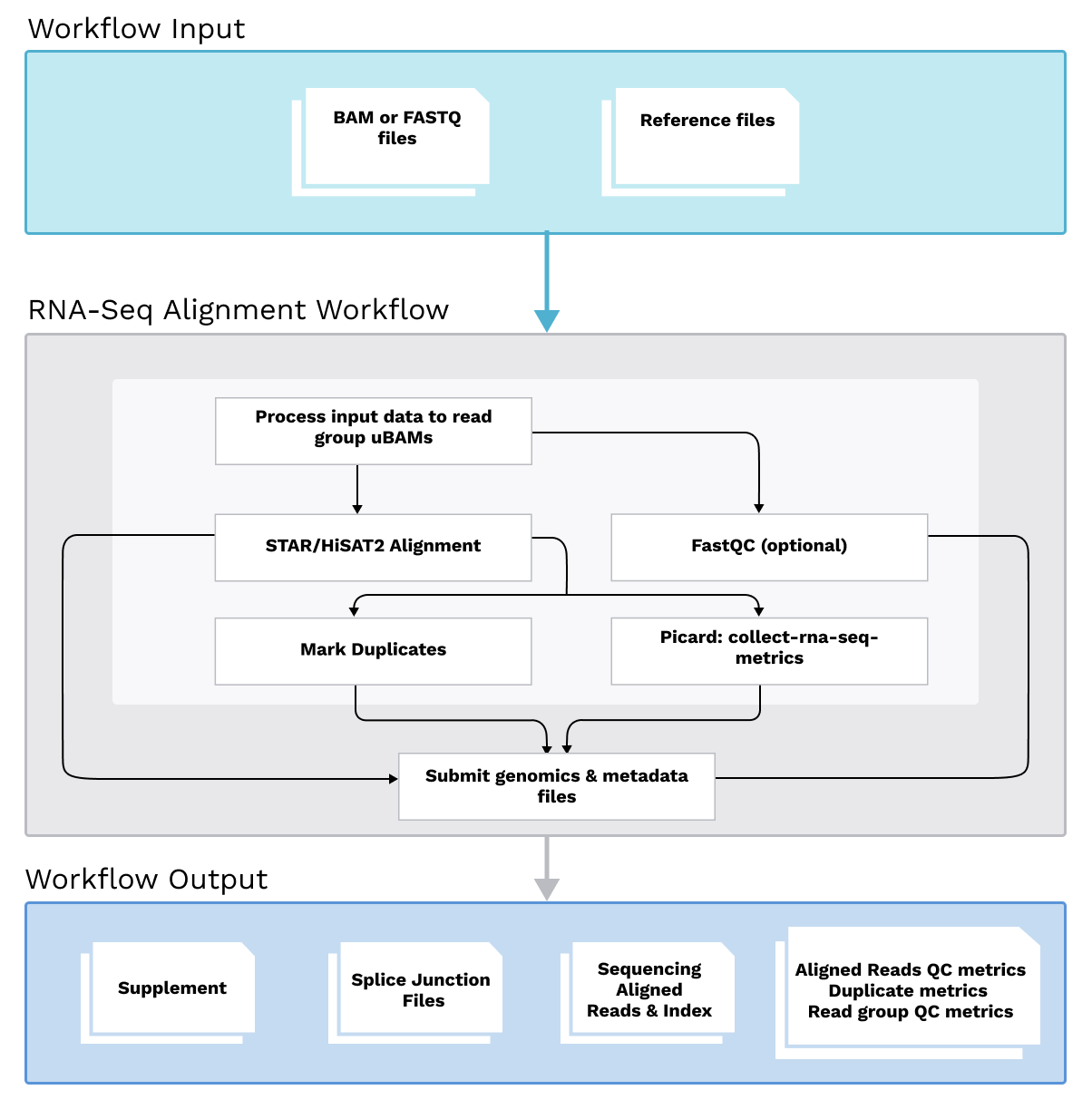
ARGO RNA-Seq Alignment workflow is a critical step in the RNA-Seq data analysis pipeline that involves mapping short RNA sequencing reads to a reference genome or transcriptome. The workflow accepts raw sequencing data in both FASTQ and BAM (aligned or unaligned) format, performs both STAR and HISAT2 alignments, and produces aligned reads in both genome and transcript coordinates, splice junctions and alignment QC reports.
Inputs
- Submitted FASTQ or BAM files(s)
- Genome Build: GRCh38_Verily_v1
- Genome Annotation: GENCODE v40
Processing
- Submitted sequencing reads (FASTQ or BAM) are converted into lane level (i.e read group level) BAMs.
- (Optional) FastQC is used for read group level Sequencing QC.
- STAR and HISAT2 are used to map the reads to the reference genome
- Biobambam2 (version 2.0.153) is used to mark duplicates per library.
- Picard:CollectRnaSeqMetrics is used to produce RNA alignment metrics
Outputs
QC metrics files
- Alignment Metrics files
- Duplicates Metrics files
Workflow Diagram