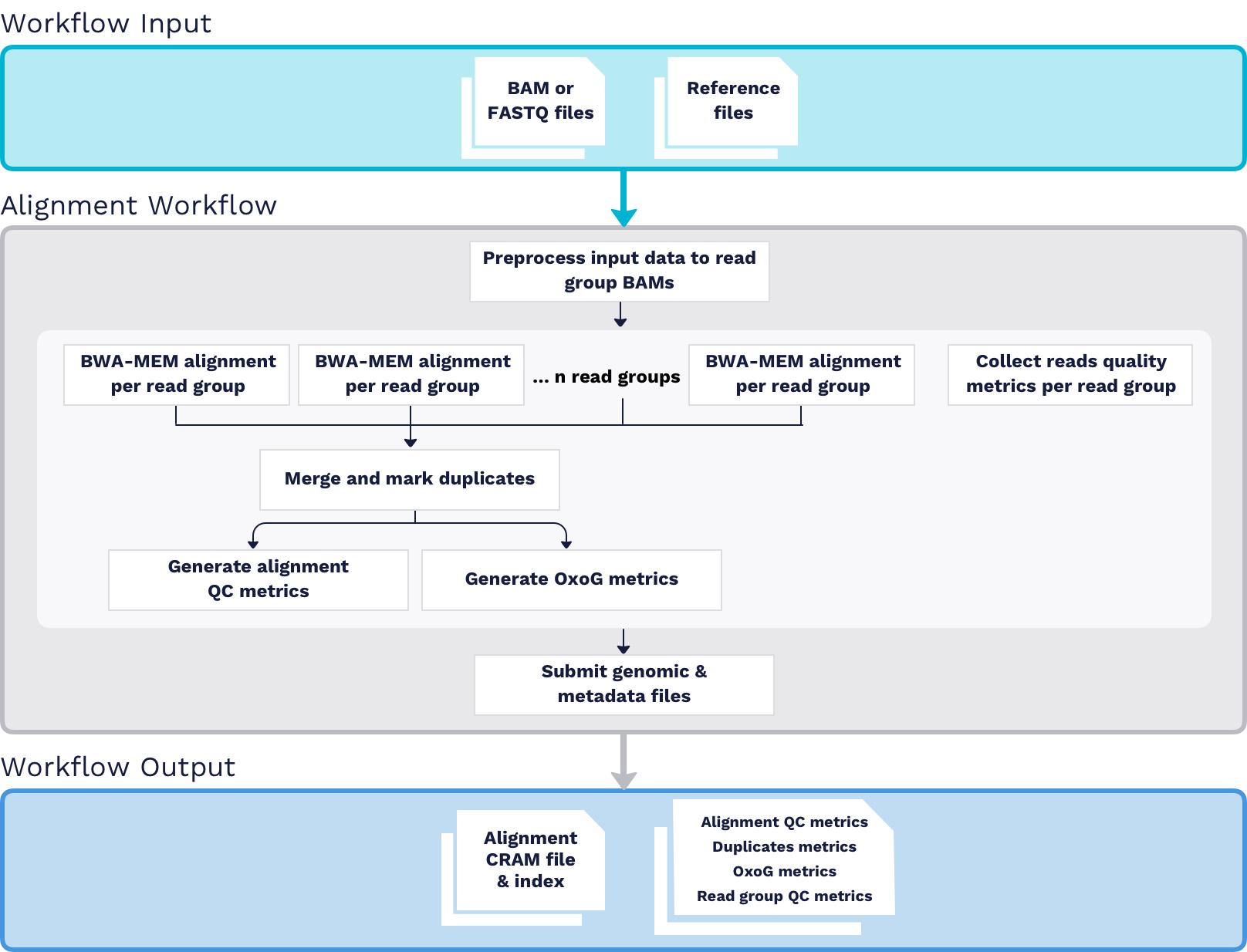
DNA-Seq Alignment
The ARGO Data Platform accepts raw sequencing data in both FASTQ and BAM (aligned or unaligned) format. The first processing step in the DNA-Seq Pipeline is uniformly aligning samples to the GRCh38 reference genome. For details, please see the latest version of the ARGO DNA-Seq Alignment.
Inputs
- All alignments are performed using GRCh38DH as the human reference genome
- Submitted FASTQ or BAM files(s)
Preprocessing
- Submitted sequencing reads (FASTQ or BAM) are converted into lane level (i.e read group level) BAMs.
- Picard:CollectQualityYieldMetrics is used for read group level BAM QC.
Processing
- BWA-MEM (version 0.7.17-r1188) is performed to map the reads to the reference genome for each read group.
- Biobambam2 (version 2.0.153) is used to merge all read group level mapped BAMs into sample level BAM and mark duplicates per library.
- Samtools:stats is used to calculate Alignment QC metrics.
- Picard:CollectOxoGMetrics is used to calculate the
OxoQscore for oxidative artifact assessment.
Outputs
- Aligned Reads CRAM and Aligned Reads Index
- QC metrics files
- Alignment Metrics files
- OxoG Metrics files
- Duplicates Metrics files
- Read Group Metrics files for each submitted read group
Workflow Diagram