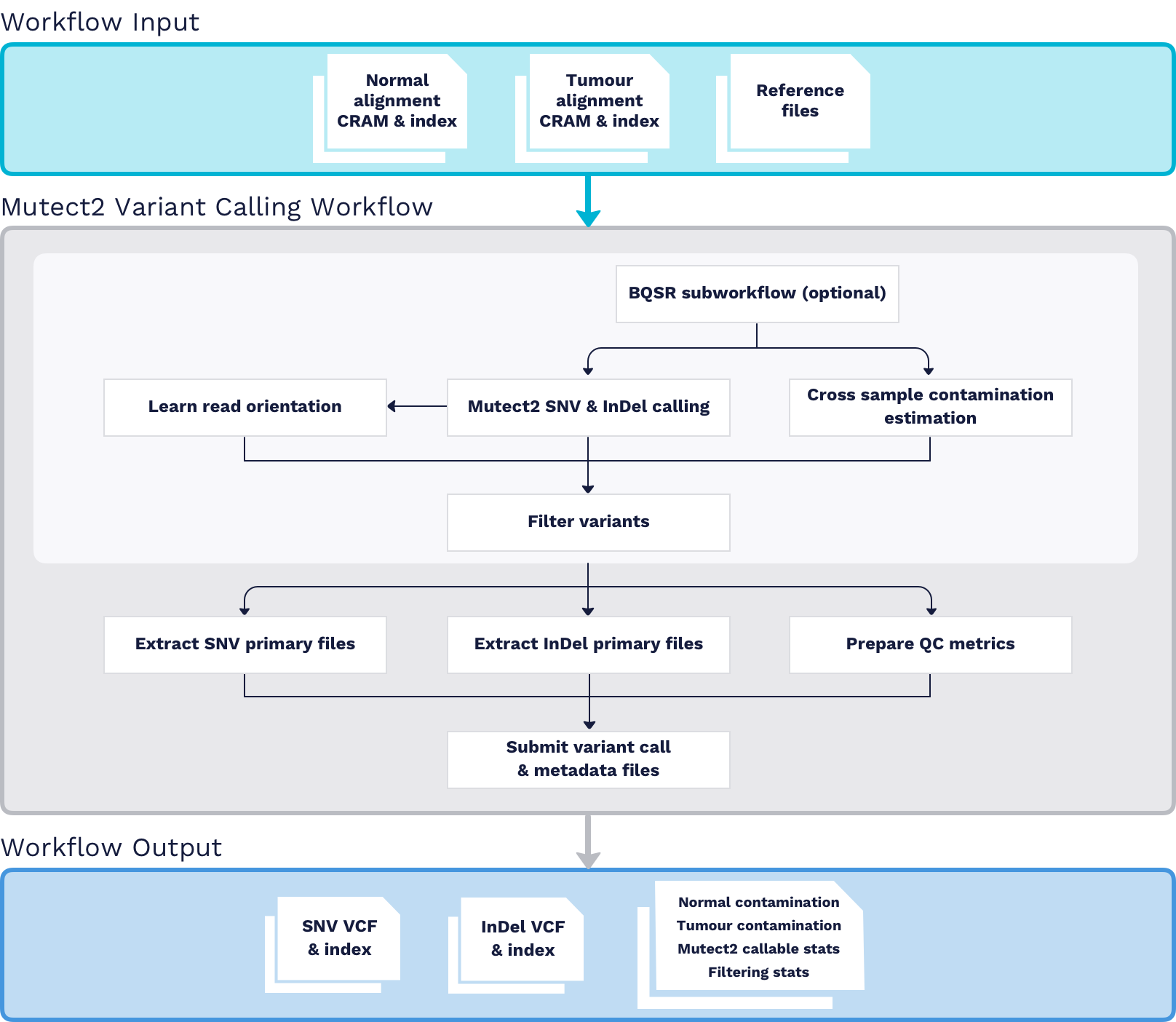
GATK Mutect2 Variant Calling
Whole genome/exome sequencing (WGS/WXS) aligned CRAM files are processed through the GATK Mutect2 Variant Calling Workflow as tumour/normal pairs. The ARGO DNA Seq pipeline has adopted the Genome Analysis Toolkit Docker Image developed at Broad Institute as the base workflow. For details, please see the latest version of the ARGO GATK Mutect2 Variant Calling workflow.
Inputs
- Normal WGS/WXS aligned CRAM and index files
- Tumour WGS/WXS aligned CRAM and index files
- Reference files
Processing
BQSR Subworkflowis an optional data pre-processing step that detects systematic errors made by the sequencing machine when it estimates the accuracy of each base call. While availble as part of the workflow, this is not run as part of the ARGO pipeline.Mutect2calls SNV and InDel simultaneously via local de-novo assembly of haplotypes in an active region.Learn Read Orientationimplements the read orientation model, which produces the --orientation-bias-artifact-priors input to the step Filter Variants.Calculate Contamination Subworkflowemits an estimate of the fraction of reads due to cross-sample contamination for both normal and tumour samples. It also generates an estimate of the allelic copy number segmentation of each tumour sample.Filter Variantsapplies filters to the raw output of Mutect2.
Collect QC Metrics
- Cross sample contamination is estimated by
GATK:CalculateContaminationfor both normal and tumour samples - Variant callable stats file is generated by
GATK:Mutect2 - Variant filtering stats file is produced by
GATK:FilterMutectCalls
Outputs
- Raw SNV Calls and VCF Index
- Raw InDel Calls and VCF Index
- QC metrics files
Workflow Diagram