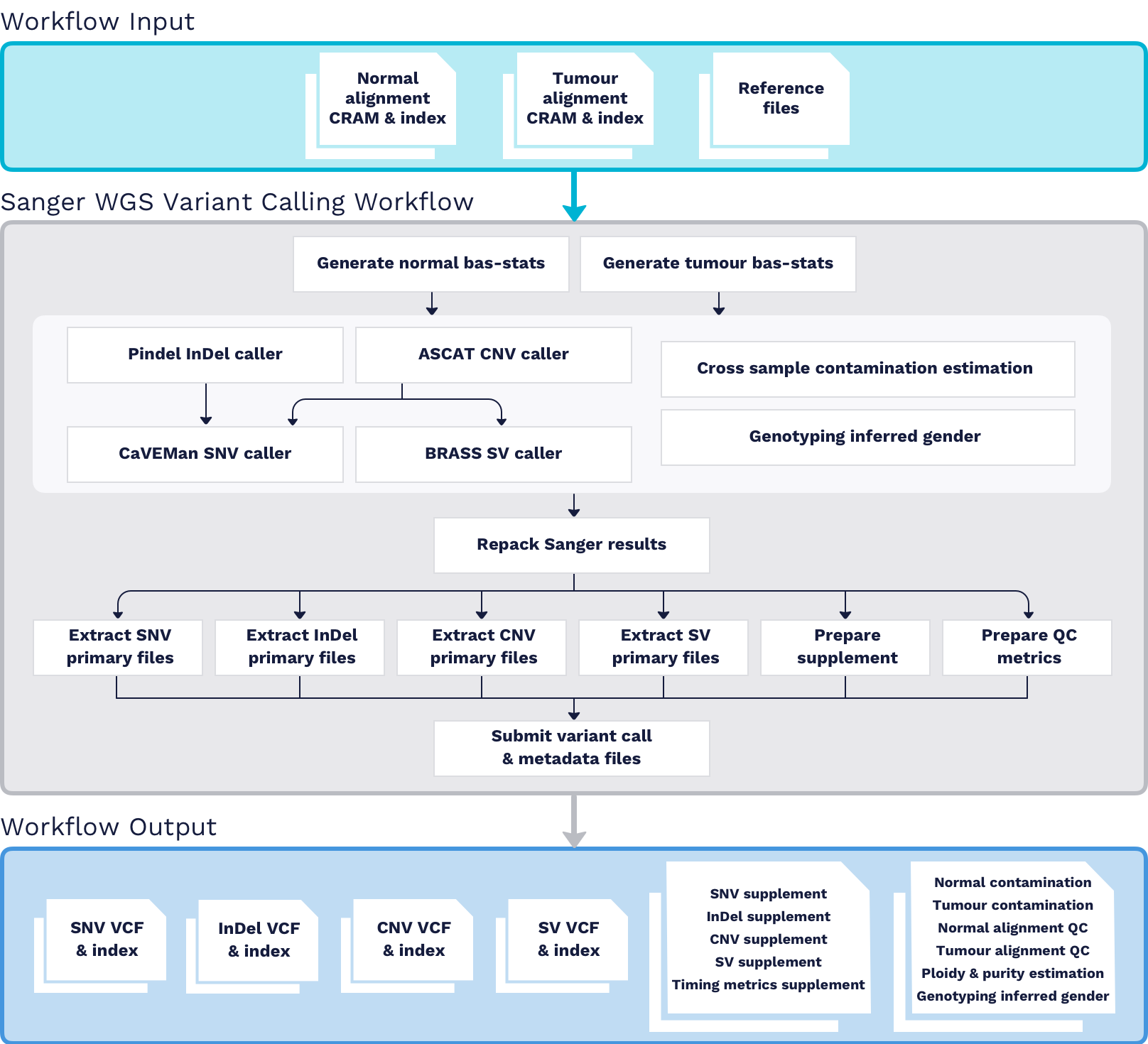
Sanger WGS Variant Calling
Whole genome sequencing (WGS) aligned CRAM files are processed through the Sanger WGS Variant Calling Workflow as tumour/normal pairs. The ARGO DNA Seq pipeline has adopted the Sanger Whole Genome Sequencing Analysis Docker Image as the base workflow. For details, please see the latest version of the ARGO Sanger WGS Variant Calling workflow.
Inputs
- Normal WGS aligned CRAM and index files
- Tumour WGS aligned CRAM and index files
- Reference files
Processing
PindelInDel caller is used for somatic insertion/deletion variant detection.ASCATCNV caller is used for somatic copy number variant analysis.CaVEManSNV caller is used for somatic single nucleotide variant analysis.BRASSSV caller is used for somatic structural variation detection.
Collect QC Metrics
- WGS aligned reads statistics are generated by Sanger:bam_stats script. The files containing normal/tumour aligned reads statistics are further used by Pindel and BRASS callers.
- Cross sample contamination is estimated by Sanger:verifyBamHomChk script for both normal and tumour samples.
- Purity and ploidy are estimated by
ASCATCNV caller - Genotypes of CRAM files from the matched normal/tumour pair are compared and the fraction of matched genotypes are produced by Sanger:compareBamGenotypes script. It also checks if the inferred genders are matched.
Outputs
- Raw SNV Calls and VCF Index
- Raw InDel Calls and VCF Index
- Raw CNV Calls and VCF Index
- Raw SV Calls and VCF Index
- SNV Supplement files
- SV Supplement files
- CNV Supplement files
- InDel Supplement files
- QC metrics files
- Alignment Metrics for both the Tumour and Normal samples
- Ploidy and Tumour Purity
- Genotyping Stats
- Cross Sample Contamination
- Runtime Stats
Workflow Diagram