Submitting Molecular Data
Molecular data consists of raw molecular data files (e.g. sequencing reads), as well as any associated file metadata (data that describes your data).
Raw molecular data is submitted to a Regional Data Processing Centre (RDPC). RDPCs are responsible for processing your program's molecular data according to the Analysis Pipeline. If you are unsure which RDPC you should submit to, please contact the DCC.
IMPORTANT: Please make sure to upgrade the icgc-argo/argo-data-submission workflow to version
1.1.0or later.
note
Sample Registration is the first step in the data submission process. You must register samples before submitting molecular data. Please ensure that your samples are registered on the ARGO Data Platform before continuing with this step.
Molecular Data Submission Workflow
icgc-argo/argo-data-submission is a workflow designed to ease the metadata payload generation and streamline molecular data submission processes. It applies sanity checks and data validation rules early on to ensure high quality of the molecular data and accompanying metadata which are to be released through the ARGO Data Platform. Initially, it was designed to support submissions for local data, but it can also work on submitting remote data stored in EGA archives. Sequencing data of FASTQ, BAM and CRAM type files are all supported, with the exception of interleaved FASTQ files.
The workflow is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity containers making installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this workflow uses one container per process which makes it much easier to maintain and update software dependencies. Here you can download the latest version.
Workflow Diagram
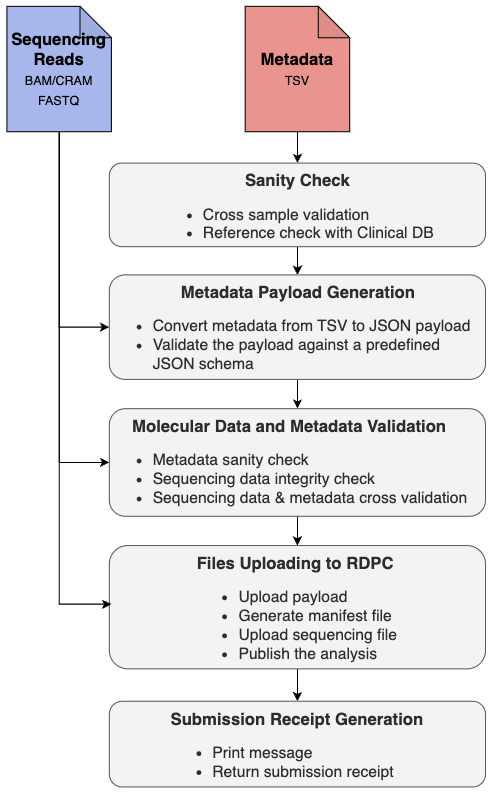
Workflow Summary
By default, it is assumed that Sequencing Reads are submitted from local storage. However if the Sequencing Reads are stored remotely in EGA Archives, it is easy to enable EGA download sub-workflow to support getting data from EGA and submit to RDPC. The workflow also adds support to Sequencing Reads in CRAM format. For this, the reference genome is required to enable cram2bam step. Currently the workflow performs the following steps:
- Sanity Checks (
sanityCheck) - [Optional] Download data from EGA Archives (
EgaDownloadWf) - [Optional] Cram to Bam conversion (
cram2bam) - Generate Metadata Payload (
pGenExp) - Validate Molecular Data and Metadata (
valSeq) - Upload Files to RDPC (
uploadWf) - Generate submission receipt (
submissionReceipt)
Quick Start
- Install Nextflow (>=22.10.0)
- Install Docker or Singularity for full workflow reproducibility
- Download the workflow and do a dry run submission with the following commands
git clone https://github.com/icgc-argo/argo-data-submission.git
cd argo-data-submission
nextflow run -preview main.nf -profile rdpc,docker -params-file example-params.local.json --api_token YOUR_API_TOKEN
This will give you a list of processes that will be invoked with the given input params for submitting data from local storage. E.g,
N E X T F L O W ~ version 23.10.0
* PREVIEW * null [confident_mahavira] DSL2 - revision: d1e7ce6cd4
[- ] process > ArgoDataSubmissionWf:sanityCheck -
[- ] process > ArgoDataSubmissionWf:checkCramReference -
[- ] process > ArgoDataSubmissionWf:pGenExp -
[- ] process > ArgoDataSubmissionWf:valSeq -
[- ] process > ArgoDataSubmissionWf:uploadWf:songSub -
[- ] process > ArgoDataSubmissionWf:uploadWf:songMan -
[- ] process > ArgoDataSubmissionWf:uploadWf:scoreUp -
[- ] process > ArgoDataSubmissionWf:uploadWf:songPub -
[- ] process > ArgoDataSubmissionWf:submissionReceipt -
[- ] process > ArgoDataSubmissionWf:printOut -
Please verify if you’ve provided the required params to invoke the expected processes.
- Start submitting your own data following the steps in section How to Upload Molecular Data
How to Upload Molecular Data
Molecular data files are submitted in conjunction with necessary, descriptive data about molecular files and samples, called metadata. The DCC requires that metadata is submitted at the same time as the molecular data to facilitate automated downstream analysis. This metadata is one of the mandatory inputs for the data submission workflow. Before proceeding, please read the instructions on how to prepare and format the metadata according to molecular metadata model and validation rules.
Step 1. Prepare Molecular Metadata
The first step of submitting molecular data is to prepare the metadata TSV files conforming to the most recent metadata dictionary that has been defined by the DCC.
The ARGO Data Platform currently accepts the following types of genomic sequencing analyses:
- DNA-Seq data from tumour and normal paired Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WXS)
- DNA-Seq data from clinical genome Targeted Sequencing (Targeted-Seq)
- RNA-Seq data from Transcriptome Sequencing (RNA-Seq)
There are submissions for either Tumour or Normal samples, from Paired-end vs. Single-end sequencing, in either BAM, FASTQ or CRAM formats. The data submission workflow can be configured to submit either local data or remote data stored in EGA archives. Each of these scenarios has a specific set of metadata rules to follow.
Experiment
Table experiment contains data elements related to a specific experiment performed for a sample. It should only contain one experiment/row entry.
Download the template and format it according to the latest dictionary.
The following are examples of correctly formatted metadata files for each case:
- WGS
- WXS
- RNA-Seq
- Targeted-Seq
- Normal Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_wgs | TEST-CA | test_wgs | test_wgs_SP_N | test_wgs_SA_N | OICR | ILLUMINA | HiSeq 2000 | WGS | 2014-12-12 | 3 | Illumina - TruSeq DNA Nano |
- Tumour Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_wgs | TEST-CA | test_wgs | test_wgs_SP_T | test_wgs_SA_T | test_wgs_SA_N | OICR | ILLUMINA | HiSeq 2000 | WGS | 2014-12-12 | 3 | Illumina - TruSeq DNA Nano |
- Normal Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_wxs | TEST-CA | test_wxs | test_wxs_SP_N | test_wxs_SA_N | OICR | ILLUMINA | HiSeq 3000 | WXS | 2014-12-12 | 2 | Agilent - SureSelect Human All Exon V6 | primary_target_001 | capture_target_001 | Hybrid Selection | Illumina - TruSeq DNA PCR-Free Library Prep Kit |
- Tumour Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_wxs | TEST-CA | test_wxs | test_wxs_SP_N | test_wxs_SA_T | test_wxs_SA_N | OICR | ILLUMINA | HiSeq 3000 | WXS | 2014-12-12 | 2 | Agilent - SureSelect Human All Exon V6 | primary_target_001 | capture_target_001 | Hybrid Selection | Illumina - TruSeq DNA PCR-Free Library Prep Kit |
- Normal Sample (Optional)
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_rna | TEST-CA | test_rna | test_rna_SP_N | test_rna_SA_N | OICR | ILLUMINA | HiSeq 3000 | RNA-Seq | 2014-12-12 | 1 | rRNA Depletion | Illumina - TruSeq RNA Library Prep Kit | FIRST_READ_SENSE_STRAND | 9 | 90% |
- Tumour Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_rna | TEST-CA | test_rna | test_rna_SP_T | test_rna_SA_T | test_rna_SA_N | OICR | ILLUMINA | HiSeq 3000 | RNA-Seq | 2014-12-12 | 1 | rRNA Depletion | Illumina - TruSeq RNA Library Prep Kit | FIRST_READ_SENSE_STRAND | 9 | 90% |
- Normal Sample (Optional)
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_targeted | TEST-CA | test_targeted | test_targeted_SP_N | test_targeted_SA_N | OICR | ILLUMINA | HiSeq 3000 | Targeted-Seq | 2014-12-12 | 1 | Illumina - Nextera Rapid Capture Kits | primary_target_002 | capture_target_002 | 341 | 10 | Hotspot Regions | Hybrid Selection | Illumina - TruSeq DNA PCR-Free Library Prep Kit |
- Tumour Sample
| type | submitter_sequencing_experiment_id | program_id | submitter_donor_id | submitter_specimen_id | submitter_sample_id | submitter_matched_normal_sample_id | sequencing_center | platform | platform_model | experimental_strategy | sequencing_date | read_group_count | target_capture_kit | primary_target_regions | capture_target_regions | number_of_genes | gene_padding | coverage | library_selection | library_preparation_kit | library_strandedness | rin | dv200 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sequencing_experiment | exp_targeted | TEST-CA | test_targeted | test_targeted_SP_T | test_targeted_SA_T | test_targeted_SA_N | OICR | ILLUMINA | HiSeq 3000 | Targeted-Seq | 2014-12-12 | 1 | Illumina - Nextera Rapid Capture Kits | primary_target_002 | capture_target_002 | 341 | 10 | Hotspot Regions | Hybrid Selection | Illumina - TruSeq DNA PCR-Free Library Prep Kit |
note
- Fields
program_id,submitter_[donor|specimen|sample]_idareRequired, and must be the same values as in thesample_registrationtable. - Field
submitter_matched_normal_sample_idisRequiredforWGS/WXStumour samples. - Fields
submitter_sequencing_experiment_id,experimental_strategy,platformandread_group_countareRequiredfor all samples. - Fields
target_capture_kit,primary_target_regionsandcapture_target_regionsareRequiredforTargeted-Seq/WXSsamples. - Field
library_strandednessisRequiredforRNA-Seqsamples.
Read_groups
Table read_groups contains data elements related to sequencing runs from an NGS experiment. Each row will correspond with a read_group. The total number of rows should match the value of field “read_group_count” in experiment.tsv
Download the template and format it according to the latest dictionary.
The following are examples of correctly formatted metadata files for each case:
- BAM/CRAM (paired)
- BAM/CRAM (single)
- FASTQ (paired)
- FASTQ (single)
| type | submitter_sequencing_experiment_id | submitter_read_group_id | read_group_id_in_bam | platform_unit | is_paired_end | file_r1 | file_r2 | library_name | read_length_r1 | read_length_r2 | insert_size | sample_barcode |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| read_group | exp_01 | C0HVY.2 | C0HVY.2 | 74_8a | true | test_rg_3.bam | test_rg_3.bam | Pond-147580 | 150 | 150 | 298 | AGCTTACC |
| read_group | exp_01 | D0RE2.1 | D0RE2.1 | 74_8b | true | test_rg_3.bam | test_rg_3.bam | Pond-147580 | 150 | 150 | 298 | AGCTTACC |
| read_group | exp_01 | D0RH0.2 | D0RH0.2 | 74_8c | true | test_rg_3.bam | test_rg_3.bam | Pond-147580 | 150 | 150 | 298 | AGCTTACC |
| type | submitter_sequencing_experiment_id | submitter_read_group_id | read_group_id_in_bam | platform_unit | is_paired_end | file_r1 | file_r2 | library_name | read_length_r1 | read_length_r2 | insert_size | sample_barcode |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| read_group | exp_02 | C0HVY.2 | C0HVY.2 | 74_8a | false | test_rg_3.cram | Pond-147580 | 150 | AGCTTACC | |||
| read_group | exp_02 | D0RE2.1 | D0RE2.1 | 74_8b | false | test_rg_3.cram | Pond-147580 | 150 | AGCTTACC | |||
| read_group | exp_02 | D0RH0.2 | D0RH0.2 | 74_8c | false | test_rg_3.cram | Pond-147580 | 150 | AGCTTACC |
| type | submitter_sequencing_experiment_id | submitter_read_group_id | read_group_id_in_bam | platform_unit | is_paired_end | file_r1 | file_r2 | library_name | read_length_r1 | read_length_r2 | insert_size | sample_barcode |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| read_group | exp_03 | C0HVY.2 | 74_8a | true | C0HVY.2_r1.fq.gz | C0HVY.2_r2.fq.gz | Pond-147580 | 150 | 150 | 298 | AGCTTACC | |
| read_group | exp_03 | D0RE2.1 | 74_8b | true | D0RE2.1_r1.fq.gz | D0RE2.1_r2.fq.gz | Pond-147580 | 150 | 150 | 298 | AGCTTACC | |
| read_group | exp_03 | D0RH0.2 | 74_8c | true | D0RH0.2_r1.fq.gz | D0RH0.2_r2.fq.gz | Pond-147580 | 150 | 150 | 298 | AGCTTACC |
| type | submitter_sequencing_experiment_id | submitter_read_group_id | read_group_id_in_bam | platform_unit | is_paired_end | file_r1 | file_r2 | library_name | read_length_r1 | read_length_r2 | insert_size | sample_barcode |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| read_group | exp_04 | C0HVY.2 | 74_8a | false | C0HVY.2.fq.gz | Pond-147580 | 150 | AGCTTACC | ||||
| read_group | exp_04 | D0RE2.1 | 74_8b | false | D0RE2.1.fq.gz | Pond-147580 | 150 | AGCTTACC | ||||
| read_group | exp_04 | D0RH0.2 | 74_8c | false | D0RH0.2.fq.gz | Pond-147580 | 150 | AGCTTACC |
note
- Fields
submitter_sequencing_experiment_id,submitter_read_group_id,platform_unit,is_paired_end, andlibrary_nameareRequiredfor all read_groups. - Field
read_group_id_in_bamisRequiredforBAM/CRAMfiles, however this can NOT be submitted for FASTQ files. - Field
file_r1isRequired, and bothfile_r1andfile_r2must match a fileName identified in thefilessection. - Fields
file_r2,read_length_r2andinsert_sizeareRequiredif and only if paired-end sequencing was done.
Files
Table files contains data elements related to the submitted files. Each row will correspond with a file object.
Download the template and format it according to the latest dictionary.
The following are examples of correctly formatted metadata files for each case:
- Local files
- EGA/Aspera
- EGA/Pyega3
| type | name | format | size | md5sum | path | ega_file_id | ega_dataset_id | ega_run_id | ega_sample_id | ega_experiment_id |
|---|---|---|---|---|---|---|---|---|---|---|
| file | test_rg_3.bam | BAM | 14245 | 178f97f7b1ca8bfc28fd5586bdd56799 | input/test_rg_3.bam |
| type | name | format | size | md5sum | path | ega_file_id | ega_dataset_id | ega_run_id | ega_sample_id | ega_experiment_id |
|---|---|---|---|---|---|---|---|---|---|---|
| file | EGAR00002324607_SLX-18928.UDP0002.bam | BAM | 48300 | 597d50657b3d171953d518fb3e02bb7a | EGAD00001007785/PART_09/EGAR00002324607_SLX-18928.UDP0002.bam.c4gh | EGAF00004257597 | EGAD00001007785 | EGAR00002324607 | EGAN00002634611 | EGAX00002220414 |
| type | name | format | size | md5sum | path | ega_file_id | ega_dataset_id | ega_run_id | ega_sample_id | ega_experiment_id |
|---|---|---|---|---|---|---|---|---|---|---|
| file | EGAR00002324607_SLX-18928.UDP0002.bam | BAM | 48300 | 597d50657b3d171953d518fb3e02bb7a | EGAF00004257597 | EGAD00001007785 | EGAR00002324607 | EGAN00002634611 | EGAX00002220414 |
note
- Fields
name,format,size,md5sumareRequiredfor all files. - Field
pathisRequiredfor both local data (use the file path relative to the directory you run the workflow) and data downloaded from EGA through Aspera (use the file path relative to Aspera root directory). - Field
ega_file_idisRequiredfor data downloaded from EGA.
Step 2. Run the Data Submission Workflow
Once you have formatted the metadata TSV files correctly, use the icgc-argo/argo-data-submission workflow to submit your data. You can get YOUR_API_TOKEN by following the instructions
Submit Local Data
- Submitting Local BAM/FASTQ files
- Submitting Local CRAM files
nextflow run main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode local \
--experiment_info_tsv path/to/experiment.tsv \
--read_group_info_tsv path/to/read_groups.tsv \
--file_info_tsv path/to/files.tsv \
--api_token YOUR_API_TOKEN
note
- Field
pathin tablefiles.tsvisRequiredfor local data and it will have to be formatted using the file path relative to the directory you run the data submission workflow - If data is submitted through a laptop, users may encounter hardware resource limitations. Memory and cpu usage can be set by adding the flags
--score_mem 16 --score_cpus 2into the nextflow command. - To run docker on ARM64 systems, users can execute
export DOCKER_DEFAULT_PLATFORM=linux/amd64in the terminal before executing the nextflow pipleline.
nextflow run main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode local \
--experiment_info_tsv path/to/experiment.tsv \
--read_group_info_tsv path/to/read_groups.tsv \
--file_info_tsv path/to/files.tsv \
--api_token YOUR_API_TOKEN \
--ref_genome_fa path/to/ref_genome_fa
note
- Provide the
ref_genome_fainfastaorfasta.gzformats if your data contains CRAMs. - This will perform a local conversion of CRAM files to BAM for submission. Please make sure you have enough local space for the converted intermediate BAM files.
- Intermediate BAM files will be deleted at the end of the pipeline process.
- Field
pathin tablefiles.tsvisRequiredfor local data and it will have to be formatted using the file path relative to the directory you run the data submission workflow
Submit Remote Data Stored in EGA Archives
If your data is hosted on EGA Archives, the pipeline provides two methods for you to download the data from EGA, then resubmit them to ARGO.
- Download data from EGA by Aspera
- Download data from EGA by Pyega3
nextflow run main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode aspera \
--experiment_info_tsv path/to/experiment.tsv \
--read_group_info_tsv path/to/read_groups.tsv \
--file_info_tsv path/to/files.tsv \
--ascp_scp_host ascp_scp_host \
--ascp_scp_user ascp_scp_user \
--aspera_scp_pass aspera_scp_pass \
--c4gh_pass_phrase c4gh_pass_phrase \
--c4gh_secret_key c4gh_secret_key \
--api_token YOUR_API_TOKEN
note
- You need to contact with EGA helpdesk to setup the aspera dbox containing your datasets, and get the credentials for
ascp_scp_host,ascp_scp_user,aspera_scp_pass - Since data present in your dbox is encrypted, you will be requested to provide EGA with an
alternative email address to get the decryption key
c4gh_pass_phrase. - The
c4gh_secret_keywill be a file found within your Aspera dbox repository. - Field
pathin tablefiles.tsvisRequiredto run with this mode and it will have to be formatted using the file path relative to Aspera root directory. - Field
ega_file_idin tablefiles.tsvisRequiredto run with this mode. Please retrieve the EGAF IDs from https://ega-archive.org/ or the EGA API. - You do NOT need to install the aspera client and crypt4gh at your local to run the data submission workflow, however you will need to login your aspera dbox to retrieve the files’ relative path and get the
c4gh_secret_key.
nextflow run main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode pyega3 \
--experiment_info_tsv path/to/experiment.tsv \
--read_group_info_tsv path/to/read_groups.tsv \
--file_info_tsv path/to/files.tsv \
--pyega3_ega_user pyega3_ega_user \
--pyega3_ega_pass pyega3_ega_pass \
--api_token YOUR_API_TOKEN
note
- Get your EGA username and password:
pyega3_ega_userandpyega3_ega_pass. Verify your credentials on https://ega-archive.org/. - Field
ega_file_idin tablefiles.tsvisRequiredto run with this mode. Please retrieve the EGAF IDs from https://ega-archive.org/ or the EGA API.
Step 3. Verify Submitted Data
Once your data has been successfully submitted through the above data submission workflow, you will receive a message providing the following information about your submitted data.
- Payload JSON File
- Analysis ID
- Submission TSV Receipt
This indicates that your data has been submitted successfully and will be queued for downstream molecular data processing.
On the other hand, you can also verify your submitted data on your Program Dashboard by viewing the card Donor Data Summary. It should display an increase in the number of Raw Reads corresponding to the submitted sample in the appropriate Tumour/Normal column.
note
- There are different tabs according to DNA-Seq and RNA-seq respectively.
- There are some delays between the submitted data that are viewable on the Program Dashboard and the completion of your submission. Please contact the DCC admin if the delay persists after 24 hours.
Understanding the Metadata Model
Graph Representation
The molecular metadata model is represented as a graph with nodes and edges. It maintains the critical relationship between entities including program, donor, specimen, sample, experiment, read groups and files. The node/edge structure is depicted below:
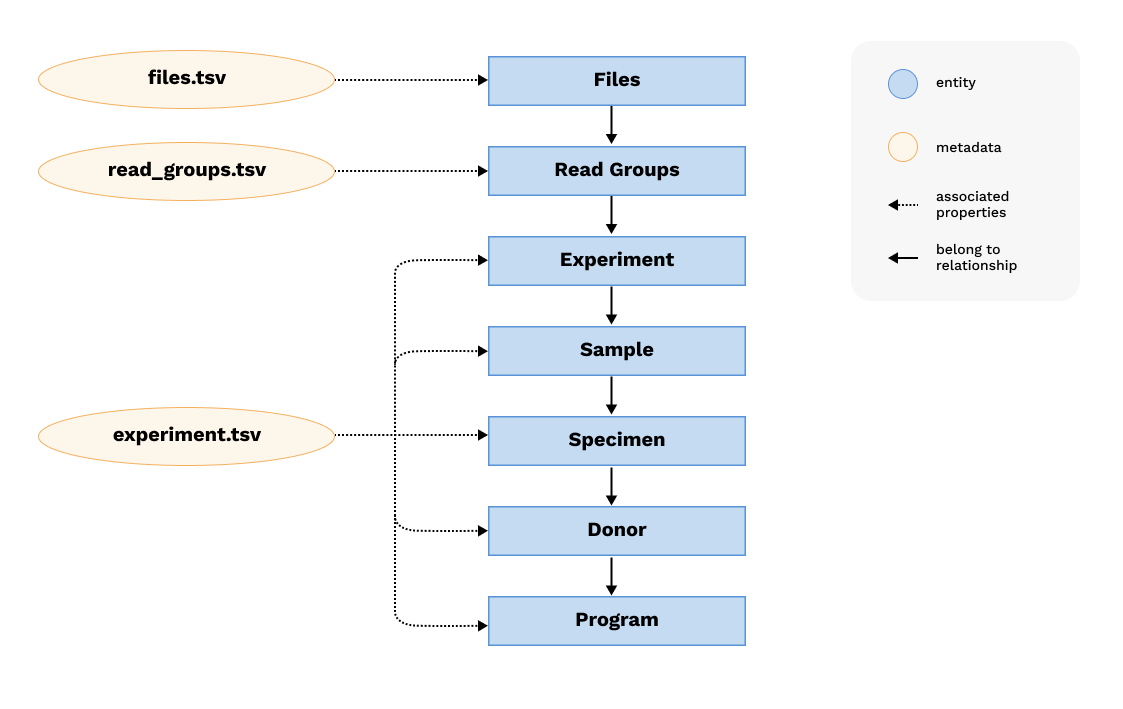
Each blue node represents an entity, edges between nodes represent relationships between entities. There is a lot of metadata associated with entities, which are represented by yellow nodes.
Metadata Dictionary
The following describes the attributes and permissible values for all of the fields within the metadata tsv files for the ARGO Data Platform and Molecular Data Processing.
- Experiment
- Read_groups
- Files
| Field | Attribute | Description | Permissible Values | Note |
|---|---|---|---|---|
| type |  | table type | sequencing_experiment | |
| submitter_sequencing_experiment_id | | Unique identifier of the sequencing experiment, assigned by the data provider. | String values that meet the regular expression ^[A-Za-z0-9\-\.\\_]{1,98}\$ | |
| program_id | | ARGO Program ID, the unique identifier of your program. If you have logged into the ARGO Data Platform, this is the Program ID that you see in the Program Services area. | ||
| submitter_donor_id | | Unique identifier of the donor, assigned by the data provider. | Values must meet the regular expression ^[A-Za-z0-9-.\\_]{1,64}\$ | Must be the same value as in sample_registration table |
| submitter_specimen_id | | Unique identifier of the specimen, assigned by the data provider. | Values must meet the regular expression ^[A-Za-z0-9-.\\_]{1,64}\$ | Must be the same value as in sample_registration table |
| submitter_sample_id | | Unique identifier of the sample, assigned by the data provider. If submitted along with BAM molecular data, must also be present in header SM. | Values must meet the regular expression ^[A-Za-z0-9-.\\_]{1,64}\$ | Must be the same value as in sample_registration table |
| submitter_matched_normal_sample_id |  | Provide the identifier of matched normal sample used for data analysis. | Values must meet the regular expression ^[A-Za-z0-9-._]{1,64}\$ or empty(null) | Required for WGS/WXS tumour samples |
| read_group_count | | The number of read groups in the molecular files being submitted. | A minimum of 1 is required. | |
| platform | | The sequencing platform type used in data generation. | CAPILLARY, LS454, ILLUMINA, SOLID, HELICOS, IONTORRENT, ONT, PACBIO, Nanopore, BGI | |
| experimental_strategy | | The primary experimental method. For sequencing data it refers to how the sequencing library was made. | WGS, WXS, RNA-Seq, Bisulfite-Seq, ChIP-Seq, Targeted-Seq | |
| sequencing_date | Optional | The date of sequencing | datetime format, for example: 2019-06-16 or 2019-06-16T20:20:39+00:00 or empty(null) | |
| platform_model | Optional | The model number of the sequencing machine used in data generation. | Any string value or empty(null) | |
| sequencing_center | Optional | Data centre sequencing was performed. Can also be specified with BAM header CN. | Any string value or empty(null) | |
| target_capture_kit | | Description that can uniquely identify a target capture kit. Suggested value is a combination of vendor, kit name, and kit version. | Any string value or empty(null) | Required for Targeted-Seq/WXS |
| primary_target_regions | | A bed file which holds the biologically relevant target regions (based on a genome, e.g. GRCh38) to capture by the assay. | ⚡ Customized Enum values which can be mapped to fileName and fileURL maintained by DCC | Required for Targeted-Seq/WXS |
| capture_target_regions | | A bed file which holds the technically relevant probes regions to capture by the assay. | ⚡ Customized Enum values which can be mapped to fileName and fileURL maintained by DCC | Required for Targeted-Seq/WXS |
| number_of_genes | Optional | Number of genes the assay is targeting | Integer with a minimum value of 1 or empty(null). | Optional for Targeted-Seq |
| gene_padding | Optional | Number of basepairs to add to exon endpoints for the inBED filter | Integer with a minimum value of 0 or empty(null). | Optional for Targeted-Seq |
| coverage | Optional | List of coverage | Hotspot Regions, Coding Exons, Introns, Promoters, or empty(null) | Optional for Targeted-Seq |
| library_selection | Optional | The method used to select and/or enrich the material being sequenced. | Affinity Enrichment, Hybrid Selection, miRNA Size Fractionation, Other, PCR, Poly-T Enrichment, Random, rRNA Depletion, or empty(null) | Optional for WXS, Targeted-Seq and RNA-Seq |
| library_preparation_kit | Optional | Provide the kit information being used for library construction. Suggested value is a combination of vendor, kit name, and kit version. | String or empty(null) | |
| library_strandedness | | Indicate the library strandedness | UNSTRANDED, FIRST_READ_SENSE_STRAND, FIRST_READ_ANTISENSE_STRAND, or empty(null) | Required for RNA-Seq |
| rin | Optional | A numerical assessment of the integrity of RNA based on the entire electrophoretic trace of the RNA sample including the presence or absence of degradation products. | A number between 1 to 10 or empty(null) | Optional for RNA-Seq |
| dv200 | Optional | The percentage of RNA fragments that are >200 nucleotides in size | A percentage, for example: 95% or empty(null) | Optional for RNA-Seq |
note
⚡ Please contact DCC to create the Customized Enum values. ⚡
| Field | Attribute | Description | Permissible Values | Note |
|---|---|---|---|---|
| type | Required | type of the table | read_group | |
| submitter_sequencing_experiment_id | | Unique identifier of the sequencing experiment, assigned by the data provider. | String values that meet the regular expression ^[a-zA-Z0-9]{1}[a-zA-Z0-9\-\\\\\_\.:']{0,98}[a-zA-Z0-9]{1}\$ | |
| submitter_read_group_id | | The identifier of a read group. Must be unique within each sample. After submission, the submitter_read_group_id in the metadata will be used for all future @RG ID in the header. | String values that meet the regular expression ^[a-zA-Z0-9\-_:\.']+\$. | |
| read_group_id_in_bam | | Conditional required field indicating the @RD ID in the BAM header and RG:Z in BAM body. If submitted, this will be used to map the @RG ID in the BAM header to the submitter_read_group_id in the payload. | String value must meet the regular expression ^[a-zA-Z0-9\-_:\.']+\$ or empty(null). | Required for BAM/CRAM files. This can NOT be submitted for FASTQ files. |
| platform_unit | | Unique identifier indicates a particular flow cell, lane and sample/library-specific identifier. For non-multiplex sequencing, platform unit and read group have a one-to-one relationship. | Any string value. | |
| is_paired_end | | Indicate if paired-end sequencing was performed. | true, false | |
| file_r1 | | Name of the sequencing file containing reads from the first end of a sequencing run. | Any string value. | Must match a name identified in the files section. |
| file_r2 | | Name of the sequencing file containing reads from the second end of a paired-end sequencing run. Required if and only if paired-end sequencing was done. | Any string value or empty(null). | Required only for paired-end sequencing. Must match a name identified in the files section if provided |
| library_name | | Name of a sequencing library. | Any string value. | |
| read_length_r1 | Optional | Length of sequencing reads in file_r1; this corresponds to the number of sequencing cycles of the first end. | Integer with a minimum value of 20 or empty(null). | |
| read_length_r2 | Optional | Length of sequencing reads in file_r2; this corresponds to the number of sequencing cycles of the second end. | Integer with a minimum value of 20 or empty(null). | Can be populated only for paired-end sequencing. |
| insert_size | Optional | For paired-end sequencing, the average size of sequences between two sequencing ends. | Integer with a minimum value of 0 or empty(null). | Can be populated only for paired-end sequencing. |
| sample_barcode | Optional | According to the SAM specification, this is the expected barcode bases as read by the sequencing machine in the absence of errors. | Any string value or empty(null). |
| Field | Attribute | Description | Permissible Values | Note |
|---|---|---|---|---|
| type | | table type | sequencing_file | |
| name | | Name of the file. | String values must meet the regular expression ^[A-Za-z0-9_\.\-\[\]\(\)]+\$. No paths are allowed in the file name. | |
| format | | Data format of sequencing files. | BAM, FASTQ,CRAM | |
| size | | Size of the file, in bytes. | Integer with a minimum value of 0 | |
| md5sum | | Computed md5sum of the file. | String values must meet the regular expression ^[a-fA-F0-9]{32}\$ | |
| path | | The path to the file to be submitted | String or empty(null) | Required for local data (use the file path relative to the directory you run the data submission workflow) and data downloaded from EGA through Aspera (use the file path relative to Aspera dbox root directory) |
| ega_file_id | | EGA File Unique Accession ID | ^EGAF[0-9]{1,32}\$ | Required for data downloaded from EGA |
| ega_dataset_id | Optional | EGA Dataset Accession ID | ^EGAD[0-9]{1,32}\$ | |
| ega_experiment_id | Optional | EGA Experiment ID | ^EGAX[0-9]{1,32}\$ | |
| ega_sample_id | Optional | EGA Sample Accession ID | ^EGAN[0-9]{1,32}\$ | |
| ega_study_id | Optional | EGA Study Accession ID | ^EGAS[0-9]{1,32}\$ | |
| ega_run_id | Optional | EGA Run Accession ID | ^EGAR[0-9]{1,32}\$ | |
| ega_policy_id | Optional | EGA Policy Accession ID | ^EGAP[0-9]{1,32}\$ | |
| ega_analysis_id | Optional | EGA Analysis Accession ID | ^EGAZ[0-9]{1,32}\$ | |
| ega_submission_id | Optional | EGA Submission ID | ^EGAB[0-9]{1,32}\$ | |
| ega_dac_id | Optional | EGA DAC Accession ID | ^EGAC[0-9]{1,32}\$ |
Molecular Data and Metadata Validation Rules
It is very important that molecular data is submitted with valid metadata. As a helpful tool for metadata validation, DCC has developed seq-tools as a stand-alone client and incorporate it as one of the mandatory steps in the data submission workflow to run on your data before officially submitting to RDPC.
Validation will help you ensure your data is formatted correctly (with accurate identifier assignments between metadata and molecular data files) and that your submission goes smoothly. It also helps the DCC ensure that the downstream Analysis Pipelines will function seamlessly.
The seq-tools wiki lists all of the validation rules that ARGO RDPC enforces, separated by the following four categories.
- Metadata General Sanity Checks
- Sequencing Data Integrity Checks
- Sequencing Data and Metadata Cross Checks
- RNA-Seq Specific Checks
Understanding the Validation Report
Validation report are separated into five categories:
- PASS: This status indicates that the payload(s) are ready for submission.
- PASS-with-WARNING: This status indicates that the payload(s) are ready for submission, however there may be a parameter you want to double check before submission.
- PASS-with-SKIPPED-check: This status indicates that the payload(s) are ready for submission, however you've skipped the md5sum check that you may want to double check before submission.
- PASS-with-WARNING-and-SKIPPED-check: This status indicates that the payload(s) are ready for submission, however you've skipped md5sum check and there are some parameters that you may want to double check before submission.
- INVALID: This status indicates that the payload(s) are not ready for submission, and each reason for failure is listed. Errors must be fixed before you attempt to submit this payload.
Troubleshooting & FAQs
Data Validation related issues
- Error in
sanityCheck
- Unregistered Project. E.g.,
Project LUNCHTIME does not exist or no samples have been registered
This error message indicates that an incorrect project code, check if any typos occurred or verify program exists in dashboard.
- Unregistered donor/specimen/sample. E.g.,
submitter_donor_id:'BATMAN' was not found in project:'TEST-PR'. Verify sample has been registered.
This error message indicates that the required Donor-Specimen-Sample data has not been registered to the ARGO Data Platform.
- Unregistered matched normal sample. E.g.,
'submitter_matched_normal_sample_id':SubWf_tes was not found in study. Please verify 'SubWf_tes' has been registered.
This error message indicates that the required matched normal sample data has not been registered to the ARGO Data Platform.
- Project registered but not in SONG. E.g.,
Program TEST-JP does not exist in SONG. Please verify program code is correct. Otherwise contact DCC-admin for help to troubleshoot.
Since clinical and molecular metadata are managed by separate databases, upon program registration, DCC-admin will create entries into both. This error message indicates that the program is not correctly created in SONG database.
- Sample with existing analysis. E.g.,
Sample 'TEST_SUBMITTER_SAMPLE_ID_ujolwwdsmgN1'/'SA623974' has an existing published analysis '15e3ffd2-16a2-465d-a3ff-d216a2765d4f' for experiment_strategy 'WGS.'
Resubmitting an existing sample will result in the above error. Special circumstances such as replacing existing data is allowed. Please contact DCC-admin for help and instructions.
- Mismatched valid IDs. E.g.,
ID Mismatch detected. Specimen_id:'TEST_SUBMITTER_SPECIMEN_ID_ujolwwdsmgN1'/'SP223585' was not found within Donor:'DN108'/'DO263239' 's specimens
SP223585 is valid but associated with another donor. Please review Donor-Specimen-Sample data found on the ARGO Program Dashboard to make sure you are submitting the correct samples.
- Error in
pGenExp
- Missing
library_strandedness. E.g.,
'experimental_strategy' 'RNA-Seq' specified but 'library_strandedness' is missing. Resubmit with both values 'experimental_strategy' and 'library_strandedness'
Check table experiment and ensure library_strandedness is provided when experimental_strategy==RNA-Seq.
- Missing
target_capture_kit,primary_target_regionsorcapture_target_regions
'experimental_strategy' 'WXS' specified but 'target_capture_kit' is missing. Resubmit with both values 'experimental_strategy' and 'target_capture_kit'
Check table experiment and ensure target_capture_kit, primary_target_regions and capture_target_regions are all provided when experimental_strategy is Targeted-Seq or WXS.
- Missing files. E.g.,
No such file: /Users/ubuntu/Desktop/GitHub/icgc-argo/argo-data-submission/argo-data-submission-wf/tests/input/D0RH0.2_r2.fq.gz
-- Check script '/Users/ubuntu/Desktop/GitHub/icgc-argo/argo-data-submission/argo-data-submission-wf/main.nf' at line: 154 or see '.nextflow.log' file for more details
A file path specified in table file does NOT exist.
- Schema validation failure. E.g.,
Command output:
None is not of type 'boolean'
Failed validating 'type' in schema['properties']['read_groups']['items']['allOf'][0]['properties']['is_paired_end']:
{'type': 'boolean'}
On instance['read_groups'][1]['is_paired_end']:
None
Command error:
Payload failed to validate against the schema
This indicates that the field is_paired_end was not a boolean as required in the schema.
- Error in
valSeq
- Failure to validate
Payload is INVALID. Please check out details in validation report under:
/Users/esu/Desktop/GitHub/icgc-argo/argo-data-submission/argo-data-submission-wf/tests/work/31/e8eb54eb381d35b459ad943d8fbb7e
Submitted files and associated metadata did not pass validation checks. Please refer to file validation_report.INVALID.jsonl within the provided working directory to help troubleshoot.
Song/Score upload issues
- Incorrect or expired Access Token
Please follow the instructions to check your Access Token on ARGO Data Platform to verify whether it has validity and has the appropriate scope, regenerating token if needed.
- Incorrect Song/Score URLs
Check URLs for Song-Client and Score-Client to make sure they both are correct. For example the Song/Score URLs for RDPC hosted at OICR Toronto are:
- Song URL: `https://submission-song.rdpc.argo.genomeinformatics.org`
- Score URL: `https://submission-score.rdpc.argo.genomeinformatics.org`
- The Object Storage providers might be undergoing maintenance, check site banners for updates
EGA data downloading related issues
- Error in
download-aspera
Any errors or inconsistencies should be directed to EGA/maintainers of the Aspera dbox. E.g. Differing MD5 between calculated and EGA’s manifest
- Error in
decrypt-aspera
If the file does not decrypt, restart the pipeline including download. If the file is still not decrypt-able it is possible the file on the server is corrupted. Please contact the host/source of the data.
- Error in
download-pyega3
If experiencing problems with the server, please contact EGA-archive for assistance. As an alternative method, an Aspera dbox can be set up from their end.
How to resume the job
Appending a -resume on any of the aforementioned commands will resume a job e.g.
nextflow run main.nf -params-file example.json --api_token YOUR_API_TOKEN -resume
How to submit multiple samples in parallel
To efficiently handle submitting multiple samples in parallel, we suggest to run the submission workflow for each sample from different launch directory.
Assuming you have downloaded the workflow to a local directory (e.g., projectDir), you can run the submission workflow in parallel from different launch directory (e.g., launchDir) for different samples. E.g.,
cd launchDir
nextflow run path/to/projectDir/main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode local \
--experiment_info_tsv path/to/experiment.tsv \
--read_group_info_tsv path/to/read_groups.tsv \
--file_info_tsv path/to/files.tsv \
--api_token YOUR_API_TOKEN
note
- Field
pathin tablefiles.tsvisRequiredfor local data and it will have to be formatted using the file path relative to the directory you run the data submission workflow
How to submit molecular data with metadata in JSON payload instead of TSVs
If you already format the sample metadata in JSON payload, you can still run the workflow to submit your data:
nextflow run main.nf \
-profile rdpc,<singularity/docker> \
--study_id YOUR_Program_ID \
--download_mode local \
--metadata_payload_json path/to/payload.json \
--data_directory path/to/molecular_data \
--api_token YOUR_API_TOKEN \
note
- When specifying the
data_directory, it should be a path relative to the directory you run the data submission workflow. Please do Not add/at the end of the path. - Do NOT use absolute path when specifying data directory